This app is based on the gwascat R package and its ebicat38 database and shows trait-associated SNP locations of the human genome. You can visualize and compare the genomic locations of up to 8 traits simultaneously.
The National Human Genome Research Institute (NHGRI) catalog of Genome-Wide Association Studies (GWAS) is a curated resource of single-nucleotide polymorphism (SNP)-trait associations. The database contains more than 100,000 SNPs and all SNP-trait associations with a p-value <1 × 10^−5.
You can access the app at
Loading the data might take a few seconds. Patience you must have, my young padawan… ;-)
Alternatively, if you are using R, you can load the app via Github with shiny:
library(shiny)
runGitHub("ShirinG/GWAS_Shiny_App")
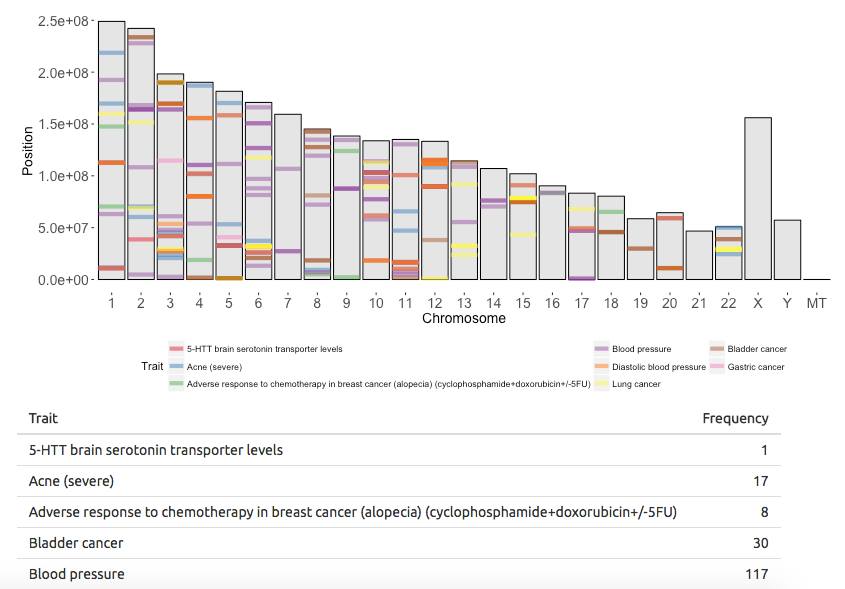
If you want to know how I built this app, contine reading.
Data preparation
Initially, I wanted to load all data directly from R packages. And while this worked in principal, it made the app load super slowly.
So, I decided to prepare the data beforehand and save the datatables as tab-delimited text files and load them in the app.
Genome information
First, I’m preparing the genome information: for each chromosome I want to know its length:
library(AnnotationDbi)
library(org.Hs.eg.db)
library(EnsDb.Hsapiens.v79)
edb <- EnsDb.Hsapiens.v79
keys <- keys(edb, keytype="SEQNAME")
chromosome_length <- select(edb, keys = keys, columns = c("SEQLENGTH", "SEQNAME"), keytype = "SEQNAME")
chromosome_length <- chromosome_length[grep("^[0-9]+$|^X$|^Y$|^MT$", chromosome_length$SEQNAME), ]
write.table(chromosome_length, "chromosome_length.txt", row.names = FALSE, col.names = TRUE, sep = "\t")
GWAS SNP data
I am saving the GWAS data as a text file as well; this datatable will be used for plotting the SNP locations.
I am also saving a table with the alphabetically sorted traits as input for the drop-down menu.
library(gwascat)
data(ebicat38)
gwas38 <- as.data.frame(ebicat38)
gwas38$DISEASE.TRAIT <- gsub("β", "beta", gwas38$DISEASE.TRAIT)
# PVALUE_MLOG: -log(p-value)
gwas38[is.infinite(gwas38$PVALUE_MLOG), "PVALUE_MLOG"] <- max(gwas38[is.finite(gwas38$PVALUE_MLOG), "PVALUE_MLOG"]) + 10
summary(gwas38[is.finite(gwas38$PVALUE_MLOG), "PVALUE_MLOG"])
summary(gwas38$PVALUE_MLOG)
# OR or BETA*: Reported odds ratio or beta-coefficient associated with strongest SNP risk allele. Note that if an OR <1 is reported this is inverted, along with the reported allele, so that all ORs included in the Catalog are >1. Appropriate unit and increase/decrease are included for beta coefficients.
summary(gwas38$OR.or.BETA)
write.table(gwas38, "gwas38.txt", row.names = FALSE, col.names = TRUE, sep = "\t")
gwas38_traits <- as.data.frame(table(gwas38$DISEASE.TRAIT))
colnames(gwas38_traits) <- c("Trait", "Frequency")
write.table(gwas38_traits, "gwas38_traits.txt", row.names = FALSE, col.names = TRUE, sep = "\t")
The Shiny App
I built my Shiny app with the traditional two-file system. This means that I have a “ui.R” file containing the layout and a “server.R” file, which contains the R code.
ui.R
From top to bottom I chose the following settings:
- loading the table with all traits
- adding a “choose below” option before the traits to have no trait chosen by default
- “united” theme for layout
- two slider bars, one for the p-value threshold and one for the odds ratio/ beta coefficient threshold
- drop-down menus for all traits, maximal eight can be chosen to be plotted simultaneously
- main panel with explanatory text, main plot and output tables
library(shiny)
library(shinythemes)
gwas38_traits <- read.table("gwas38_traits.txt", header = TRUE, sep = "\t")
diseases <- c("choose below", as.character(gwas38_traits$Trait))
shinyUI(fluidPage(theme = shinytheme("united"),
titlePanel("GWAS disease- & trait-associated SNP locations of the human genome"),
sidebarLayout(
sidebarPanel(
sliderInput("pvalmlog",
"-log(p-value):",
min = -13,
max = 332,
value = -13),
sliderInput("orbeta",
"odds ratio/ beta-coefficient :",
min = 0,
max = 4426,
value = 0),
selectInput("variable1", "First trait:",
choices = diseases[-1]),
selectInput("variable2", "Second trait:",
choices = diseases),
selectInput("variable3", "Third trait:",
choices = diseases),
selectInput("variable4", "Fourth trait:",
choices = diseases),
selectInput("variable5", "Fifth trait:",
choices = diseases),
selectInput("variable6", "Sixth trait:",
choices = diseases),
selectInput("variable7", "Seventh trait:",
choices = diseases),
selectInput("variable8", "Eighth trait:",
choices = diseases)
),
mainPanel(
br(),
p("The National Human Genome Research Institute (NHGRI) catalog of Genome-Wide Association Studies (GWAS) is a curated resource of single-nucleotide polymorphisms (SNP)-trait associations. The database contains more than 100,000 SNPs and all SNP-trait associations with a p-value <1 × 10^−5."),
p("This app is based on the 'gwascat' R package and its 'ebicat38' database and shows trait-associated SNP locations of the human genome."),
p("For more info on how I built this app check out", a("my blog.", href = "https://shiring.github.io/")),
br(),
h4("How to use this app:"),
div("Out of 1320 available traits or diseases you can choose up to 8 on the left-side panel und see their chromosomal locations below. The traits are sorted alphabetically. You can also start typing in the drop-down panel and traits matching your query will be suggested.", style = "color:blue"),
br(),
div("With the two sliders on the left-side panel you can select SNPs above a p-value threshold (-log of association p-value) and/or above an odds ratio/ beta-coefficient threshold. The higher the -log of the p-value the more significant the association of the SNP with the trait. Beware that some SNPs have no odds ratio value and will be shown regardless of the threshold.", style = "color:blue"),
br(),
div("The table directly below the plot shows the number of SNPs for each selected trait (without subsetting when p-value or odds ratio are changed). The second table below the first shows detailed information for each SNP of the chosen traits. This table shows only SNPs which are plotted (it subsets according to p-value and odds ratio thresholds).", style = "color:blue"),
br(),
div("Loading might take a few seconds...", style = "color:red"),
br(),
plotOutput("plot"),
tableOutput('table'),
tableOutput('table2'),
br(),
p("GWAS catalog:", a("http://www.ebi.ac.uk/gwas/", href = "http://www.ebi.ac.uk/gwas/")),
p(a("Welter, Danielle et al. “The NHGRI GWAS Catalog, a Curated Resource of SNP-Trait Associations.” Nucleic Acids Research 42.Database issue (2014): D1001–D1006. PMC. Web. 17 Dec. 2016.", href = "https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3965119/"))
)
)
))
server.R
This file contains the R code to produce the plot and output tables.
- The chromosome data is loaded for the chromosome length barplot. In order to have the correct order, I am setting the chromosome factor order manually
- renderPlot() contains the code for the plot
- from the eight possible input traits, all unique traits are plotted (removing “choose below” first), unique traits were set here, so as not to plot the same trait on top of themselves if the same trait is chosen in two drop-down menus
- the first output table shows the number of SNPs for the chosen traits
- the second output table shows SNP information for all SNPs of the chosen trait(s) within the chosen thresholds
library(shiny)
chr_data <- read.table("chromosome_length.txt", header = TRUE, sep = "\t")
chr_data$SEQNAME <- as.factor(chr_data$SEQNAME)
f = c("1", "2", "3", "4", "5", "6", "7", "8", "9", "10", "11", "12", "13", "14", "15", "16", "17", "18", "19", "20", "21", "22", "X", "Y", "MT")
chr_data <- within(chr_data, SEQNAME <- factor(SEQNAME, levels = f))
library(ggplot2)
gwas38 <- read.table("gwas38.txt", header = TRUE, sep = "\t")
gwas38_traits <- read.table("gwas38_traits.txt", header = TRUE, sep = "\t")
# Define server logic required to plot variables
shinyServer(function(input, output) {
# Generate a plot of the requested variables
output$plot <- renderPlot({
plot_snps <- function(trait){
if (any(trait == "choose below")){
trait <- trait[-which(trait == "choose below")]
} else {
trait <- unique(trait)
}
for (i in 1:length(unique(trait))){
trait_pre <- unique(trait)[i]
snps_data_pre <- gwas38[which(gwas38$DISEASE.TRAIT == paste(trait_pre)), ]
snps_data_pre <- data.frame(Chr = snps_data_pre$seqnames,
Start = snps_data_pre$CHR_POS,
SNPid = snps_data_pre$SNPS,
Trait = rep(paste(trait_pre), nrow(snps_data_pre)),
PVALUE_MLOG = snps_data_pre$PVALUE_MLOG,
OR.or.BETA = snps_data_pre$OR.or.BETA)
snps_data_pre <- subset(snps_data_pre, PVALUE_MLOG > input$pvalmlog)
snps_data_pre <- subset(snps_data_pre, OR.or.BETA > input$orbeta | is.na(OR.or.BETA))
if (i == 1){
snps_data <- snps_data_pre
} else {
snps_data <- rbind(snps_data, snps_data_pre)
}
}
snps_data <- within(snps_data, Chr <- factor(Chr, levels = f))
p <- ggplot(data = snps_data, aes(x = Chr, y = as.numeric(Start))) +
geom_bar(data = chr_data, aes(x = SEQNAME, y = as.numeric(SEQLENGTH)), stat = "identity", fill = "grey90", color = "black") +
theme(
axis.text = element_text(size = 14),
axis.title = element_text(size = 14),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
panel.background = element_rect(fill = "white"),
legend.position = "bottom"
) +
labs(x = "Chromosome", y = "Position")
p + geom_segment(data = snps_data, aes(x = as.numeric(as.character(Chr)) - 0.45, xend = as.numeric(as.character(Chr)) + 0.45,
y = Start, yend = Start, colour = Trait), size = 2, alpha = 0.5) +
scale_colour_brewer(palette = "Set1") +
guides(colour = guide_legend(ncol = 3, byrow = FALSE))
}
plot_snps(trait = c(input$variable1, input$variable2, input$variable3, input$variable4, input$variable5, input$variable6, input$variable7, input$variable8, input$variable9))
})
output$table <- renderTable({
table <- gwas38_traits[which(gwas38_traits$Trait %in% c(input$variable1, input$variable2, input$variable3, input$variable4, input$variable5, input$variable6, input$variable7, input$variable8)), ]
})
output$table2 <- renderTable({
table <- gwas38[which(gwas38$DISEASE.TRAIT %in% c(input$variable1, input$variable2, input$variable3, input$variable4, input$variable5, input$variable6, input$variable7, input$variable8)), c(1:5, 8, 10, 11, 13, 14, 20, 27, 32, 33, 34, 36)]
table <- subset(table, PVALUE_MLOG > input$pvalmlog)
table <- subset(table, OR.or.BETA > input$orbeta | is.na(OR.or.BETA))
table[order(table$seqnames), ]
})
})
Deploying to shinyapps.io
Finally, I am deploying my finished app to shinyapps.io with the rsconnect package. You will need to register with shinyapps.io before you can host your Shiny app there and register rsconnect with the token you received.
library(rsconnect)
rsconnect::deployApp('~/Documents/Github/blog_posts_prep/gwas/shiny/GWAS_Shiny_App')
This will open your deployed app right aways. You can now share the link to your app with the world! :-)
If you are interested in human genomics…
… you might also like these posts:
- https://shiring.github.io/genome/2016/10/23/AnnotationDbi
- https://shiring.github.io/genome/2016/11/01/AnnotationDbi_part2
- https://shiring.github.io/genome/2016/12/11/homologous_genes_post
- https://shiring.github.io/genome/2016/12/14/homologous_genes_part2_post
## R version 3.3.2 (2016-10-31)
## Platform: x86_64-apple-darwin13.4.0 (64-bit)
## Running under: macOS Sierra 10.12.1
##
## locale:
## [1] de_DE.UTF-8/de_DE.UTF-8/de_DE.UTF-8/C/de_DE.UTF-8/de_DE.UTF-8
##
## attached base packages:
## [1] parallel stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] rsconnect_0.6
## [2] gwascat_2.6.0
## [3] Homo.sapiens_1.3.1
## [4] TxDb.Hsapiens.UCSC.hg19.knownGene_3.2.2
## [5] GO.db_3.4.0
## [6] OrganismDbi_1.16.0
## [7] EnsDb.Hsapiens.v79_2.1.0
## [8] ensembldb_1.6.2
## [9] GenomicFeatures_1.26.0
## [10] GenomicRanges_1.26.1
## [11] GenomeInfoDb_1.10.1
## [12] org.Hs.eg.db_3.4.0
## [13] AnnotationDbi_1.36.0
## [14] IRanges_2.8.1
## [15] S4Vectors_0.12.1
## [16] Biobase_2.34.0
## [17] BiocGenerics_0.20.0
##
## loaded via a namespace (and not attached):
## [1] colorspace_1.3-1 rprojroot_1.1
## [3] biovizBase_1.22.0 htmlTable_1.7
## [5] XVector_0.14.0 base64enc_0.1-3
## [7] dichromat_2.0-0 base64_2.0
## [9] interactiveDisplayBase_1.12.0 codetools_0.2-15
## [11] splines_3.3.2 snpStats_1.24.0
## [13] ggbio_1.22.0 fail_1.3
## [15] doParallel_1.0.10 knitr_1.15.1
## [17] Formula_1.2-1 gQTLBase_1.6.0
## [19] Rsamtools_1.26.1 cluster_2.0.5
## [21] graph_1.52.0 shiny_0.14.2
## [23] httr_1.2.1 backports_1.0.4
## [25] assertthat_0.1 Matrix_1.2-7.1
## [27] lazyeval_0.2.0 limma_3.30.6
## [29] acepack_1.4.1 htmltools_0.3.5
## [31] tools_3.3.2 gtable_0.2.0
## [33] reshape2_1.4.2 dplyr_0.5.0
## [35] fastmatch_1.0-4 Rcpp_0.12.8
## [37] Biostrings_2.42.1 nlme_3.1-128
## [39] rtracklayer_1.34.1 iterators_1.0.8
## [41] ffbase_0.12.3 stringr_1.1.0
## [43] mime_0.5 XML_3.98-1.5
## [45] AnnotationHub_2.6.4 zlibbioc_1.20.0
## [47] scales_0.4.1 BSgenome_1.42.0
## [49] VariantAnnotation_1.20.2 BiocInstaller_1.24.0
## [51] SummarizedExperiment_1.4.0 RBGL_1.50.0
## [53] RColorBrewer_1.1-2 BBmisc_1.10
## [55] yaml_2.1.14 memoise_1.0.0
## [57] gridExtra_2.2.1 ggplot2_2.2.0
## [59] biomaRt_2.30.0 rpart_4.1-10
## [61] reshape_0.8.6 latticeExtra_0.6-28
## [63] stringi_1.1.2 RSQLite_1.1-1
## [65] foreach_1.4.3 checkmate_1.8.2
## [67] BiocParallel_1.8.1 BatchJobs_1.6
## [69] GenomicFiles_1.10.3 bitops_1.0-6
## [71] matrixStats_0.51.0 evaluate_0.10
## [73] lattice_0.20-34 GenomicAlignments_1.10.0
## [75] bit_1.1-12 GGally_1.3.0
## [77] plyr_1.8.4 magrittr_1.5
## [79] sendmailR_1.2-1 R6_2.2.0
## [81] Hmisc_4.0-1 DBI_0.5-1
## [83] foreign_0.8-67 mgcv_1.8-16
## [85] survival_2.40-1 RCurl_1.95-4.8
## [87] nnet_7.3-12 tibble_1.2
## [89] rmarkdown_1.2 grid_3.3.2
## [91] data.table_1.10.0 gQTLstats_1.6.0
## [93] digest_0.6.10 xtable_1.8-2
## [95] ff_2.2-13 httpuv_1.3.3
## [97] brew_1.0-6 openssl_0.9.5
## [99] munsell_0.4.3 beeswarm_0.2.3
## [101] Gviz_1.18.1
